
Comprehensive Amyloidosis Program
The UCSF Comprehensive Amyloidosis Program is a nationally recognized program committed to offering state-of-the-art approaches for diagnosis, management and research of amyloidosis. The Amyloidosis Program brings together UCSF Health specialists from hematology-oncology, neurology, nephrology, radiology, and pathology to offer our patients integrated diagnostic and treatment strategies. Our patients are followed closely by physicians, advanced practice providers, nurses, and pharmacists in the UCSF Advanced Heart Failure Comprehensive Care Center and Cardio-Oncology clinics to help manage symptoms of heart failure and maintain the best quality of life possible. The Amyloidosis Program is actively involved in research endeavors through institutional, national, and international collaboration.
Program Leadership

Mandar Aras, MD, PhD
Assistant Professor, Medicine and Co-Director Cardiac Amyloidosis Program
Cardiology
Patient Care

Advanced Heart Failure Comprehensive Care Center
400 Parnassus Ave., Fifth Floor
San Francisco, CA 94143
(415) 502-4243
What is Cardiac Amyloidosis?
Amyloidosis is a group of diseases characterized by protein deposition, where misfolded proteins form amyloid fibrils that deposit in organs. The accumulation of misfolded proteins in organs can cause mechanical disruption and organ dysfunction. Cardiac amyloidosis refers to when amyloid proteins build up in the heart muscle, leading the muscle to become stiff. This is referred to restrictive cardiomyopathy, and as a result your heart struggles to pump enough blood to the rest of the body.
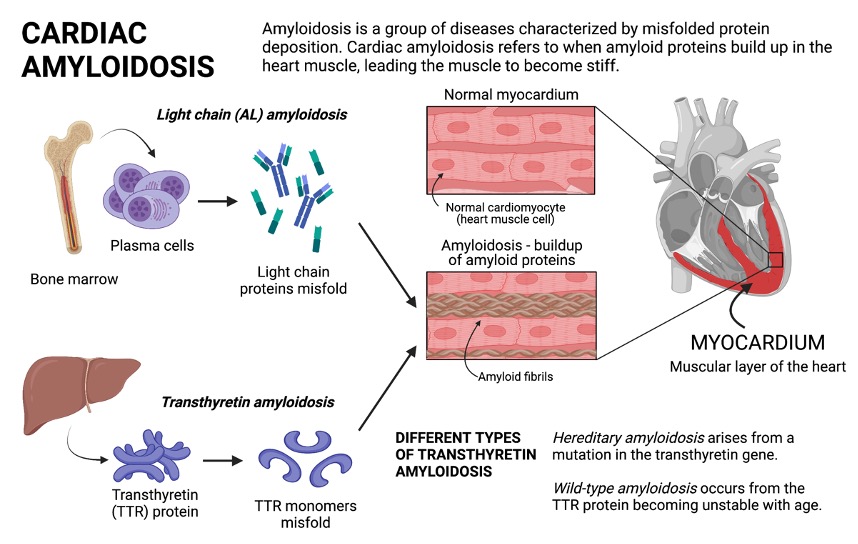
What are the Different Types of Cardiac Amyloidosis?
The major types of cardiac amyloidosis are light chain (AL) and transthyretin (TTR) amyloidosis, which are named for the proteins that cause disease.
- Light chain amyloidosis involves plasma cells from the bone marrow producing excess light chains that misfold and form clumps of amyloid fibrils. The light chain fibrils circulate and deposit in different organs, where they can cause serious damage and dysfunction.
- Transthyretin amyloidosis involves the dissociation and misfolding of the transthyretin (TTR) protein. The normal function of transthyretin is to transport thyroxine, a thyroid hormone, and retinol (vitamin A). In TTR amyloidosis, the abnormal TTR protein dissociates into individual components (monomers), which are prone to misfolding and clumping into amyloid fibrils, which then build up in different organs and cause organ dysfunction.
- There are two main forms of TTR amyloidosis. In hereditary TTR amyloidosis, patients have a genetic mutation in the transthyretin gene that results in faulty TTR proteins made in the liver. This condition can be passed onto family members. Wild-type (WT) amyloidosis results from the natural TTR protein becoming unstable with age.
What are the Signs and Symptoms of Cardiac Amyloidosis?
Amyloid deposits can build up in the heart and cause the heart muscle to become thick, which affects its ability to pump effectively. This can lead to congestive heart failure, rhythm disturbances, or conduction disorders.
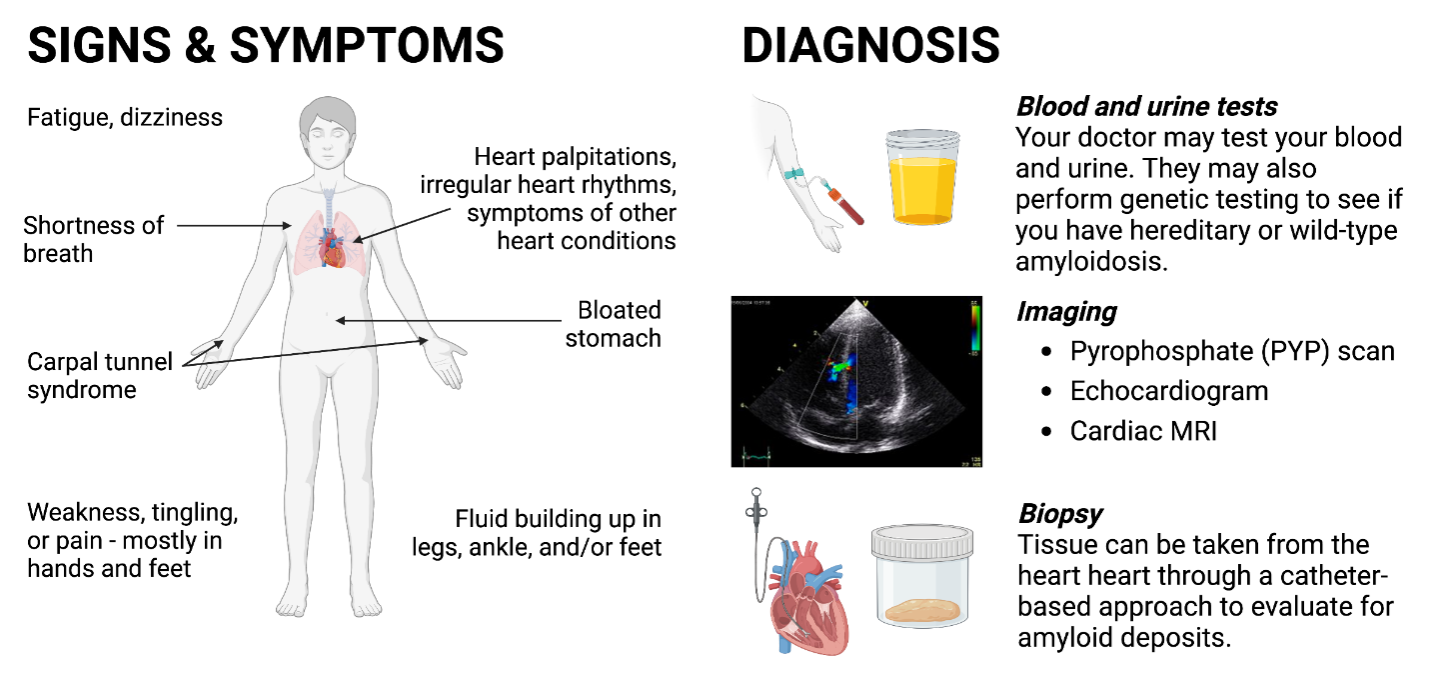
General symptoms of cardiac amyloidosis include the following:
- Shortness of breath, at rest or while doing physical activity
- Fluid retention in abdomen, legs, ankles, and/or feet
- Irregular heart rhythms (arrhythmias)
- Heart palpitations
- Fatigue
- Dizziness
There are also non-cardiac symptoms that can occur because of amyloid fibrils depositing in other parts of the body, such as:
- Carpal tunnel syndrome – often in both wrists
- Lumbar spinal stenosis – narrowing of the spinal canal, which creates pressure on the spinal cord and nerves
- Peripheral neuropathy – nerve damage causing weakness, numbness, and pain usually in the hands and feet
How is Cardiac Amyloidosis Diagnosed and Treated?
Depending on the type of amyloidosis suspected, there are several diagnostic tests possible:
- Blood and urine testing – serum free light chains and serum immunofluorescence (to evaluate for AL amyloidosis), troponin (to detect heart injury), natriuretic peptides (BNP or NTproBNP, to assess if the pressure inside your heart is increased)
- Echocardiography – to evaluate heart function and structure; a thicker heart wall may indicate presence of amyloidosis
- Pyrophosphate (PYP) scan – to evaluate presence of TTR amyloid deposits in the heart
- Cardiac magnetic resonance imaging (MRI) – to determine degree of fibrosis in the heart, and assess heart function or structure
- Cardiac Biopsy – to look for presence of amyloid deposits and fibrils; tissue can be taken from the heart
- Genetic testing – if hereditary TTR amyloidosis is suspected, genetic testing identifies the specific disease-causing mutation
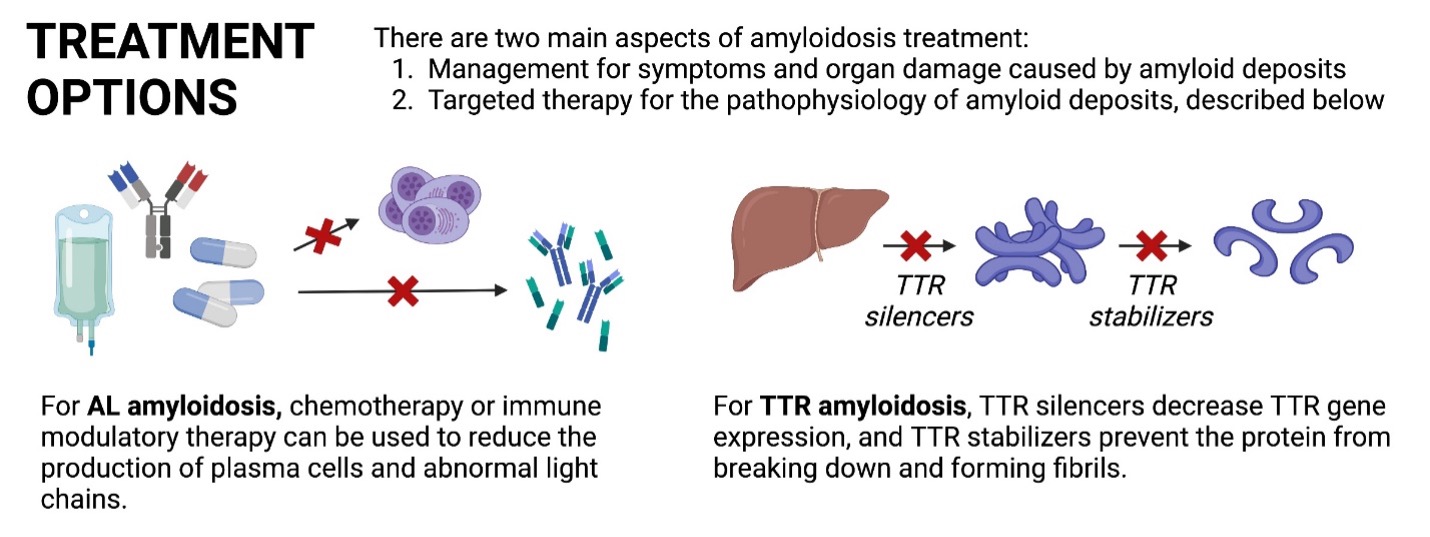
Treatment also varies depending on the type of amyloidosis. There are two aspects of amyloidosis treatment: one to treat the symptoms and organ damage caused by amyloid deposits and one to target the pathophysiology of amyloid deposition. The specific treatments to address the latter are listed below:
- AL amyloidosis: chemotherapy, immune modulatory therapy, and/or steroids can be used to reduce the production of plasma cells and abnormal light chain proteins
- TTR amyloidosis: TTR silencers decrease TTR gene expression by the liver, and TTR stabilizers prevent the TTR protein from breaking down and forming amyloid deposits.
- In some cases, stem cell transplant or heart transplantation can be considered.
Cardiac Amyloidosis Research at UCSF
At UCSF, we routinely participate in clinical trials and expanded access programs for the new and existing therapies. Here are a few of the current amyloidosis clinical trials and research projects we participate in:
- CardioTTRansform: A Study to Evaluate the Efficacy and Safety of Eplontersen in Participants With Transthyretin-Mediated Amyloid Cardiomyopathy (ATTR CM)
- Cardiac Amyloidosis Registry Study (CARS): a registry of cardiac amyloidosis patients to describe the characteristics and outcomes of patients with amyloidosis. In collaboration with Cedars Sinai Medical Center and other site
- CAEL-101: A Study to Evaluate the Efficacy and Safety of CAEL-101 in Patients With Mayo Stage IIIa AL Amyloidosis